2021-12-06
/ Last updated : 2021-12-06
adiqb1
Laboratory of Computational Genomics
HiC1Dmetrics: framework to extract various one-dimensional features from chromosome structure data
Jiankang Wang
Ryuichiro Nakato
Briefings in Bioinformatics
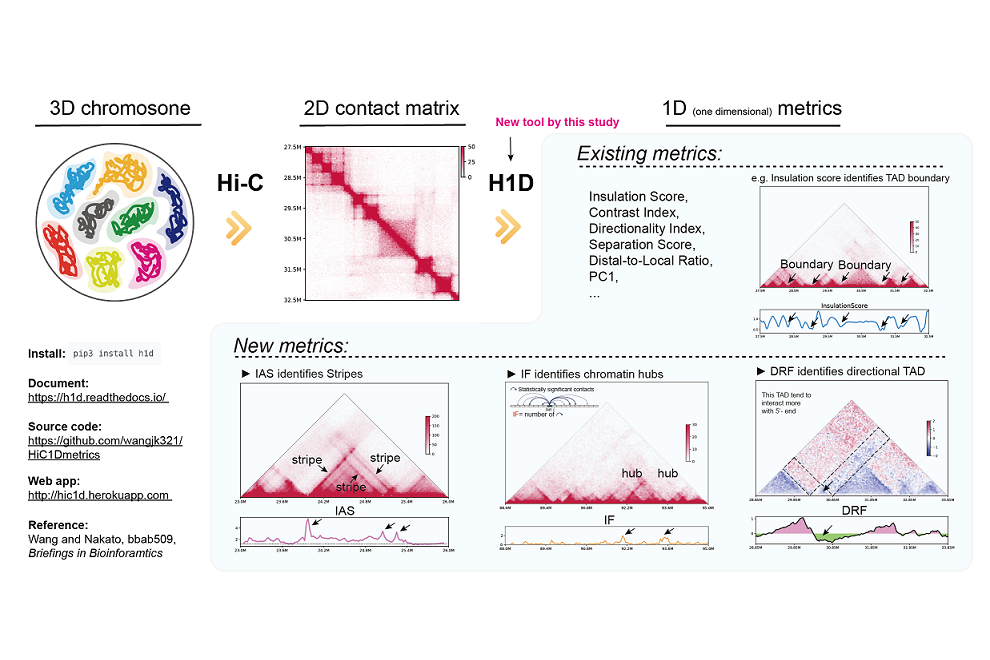
Hi-C (a high-throughput chromosome conformation capture method) is a powerful method to obtain information on hierarchical chromatin structure. The typical Hi-C analysis uses a two-dimensional (2D) contact matrix that indicates contact frequencies between all possible genomic position pairs. However, such a process is computationally expensive and not amenable to handling quantitative comparisons, visualizations, and integrations across multiple datasets.
Jiankang Wang, a Ph.D. student, and Ryuichiro Nakato, a lecturer at the Institute for Quantitative Biosciences, the University of Tokyo, developed a new method called “HiC1Dmetrics” that can extract a wide variety of one-dimensional (1D) metrics from Hi-C data (https://h1d. readthedocs.io/en/latest/index.html). They also proposed several new 1D metrics to identify additional unique chromosome structures such as stripe TADs and chromatin hubs. The experiments for human Hi-C samples showed that 1D metrics could be easily combined with epigenome tracks to annotate chromatin states in greater detail.
This tool facilitates quantitative comparison of 3D structures among multiple samples and enables fast and memory-efficient analysis of high-resolution data, which requires a large amount of time for calculation. HiC1Dmetrics is open-source (github.com/wangjk321/HiC1Dmetrics) and can be accessed from both command-line and web-based interfaces. Our tool constitutes a valuable resource for the community of chromosome-organization researchers.
Pressrelease
(Go to the IQB Japanese Website)
Journals
Journal: Briefings in Bioinformatics
Title: HiC1Dmetrics: framework to extract various one-dimensional features from chromosome structure data
Author: Jiankang Wang and Ryuichiro Nakato*
DOI: 10.1093/bib/bbab509
URL:
https://doi.org/10.1093/bib/bbab509
Contact
Ryuichiro Nakato
Lecturer, Institute for Quantitative Biosciences