Theme
Genome-wide analysis using next-generation sequencing (NGS) is a central method in genomics fields. We aim to develop a pipeline for large-scale NGS analysis and achieve an epoch-making discovery without help from existing biological knowledge.
About Research
Large-scale data-driven analysis for computational genomics
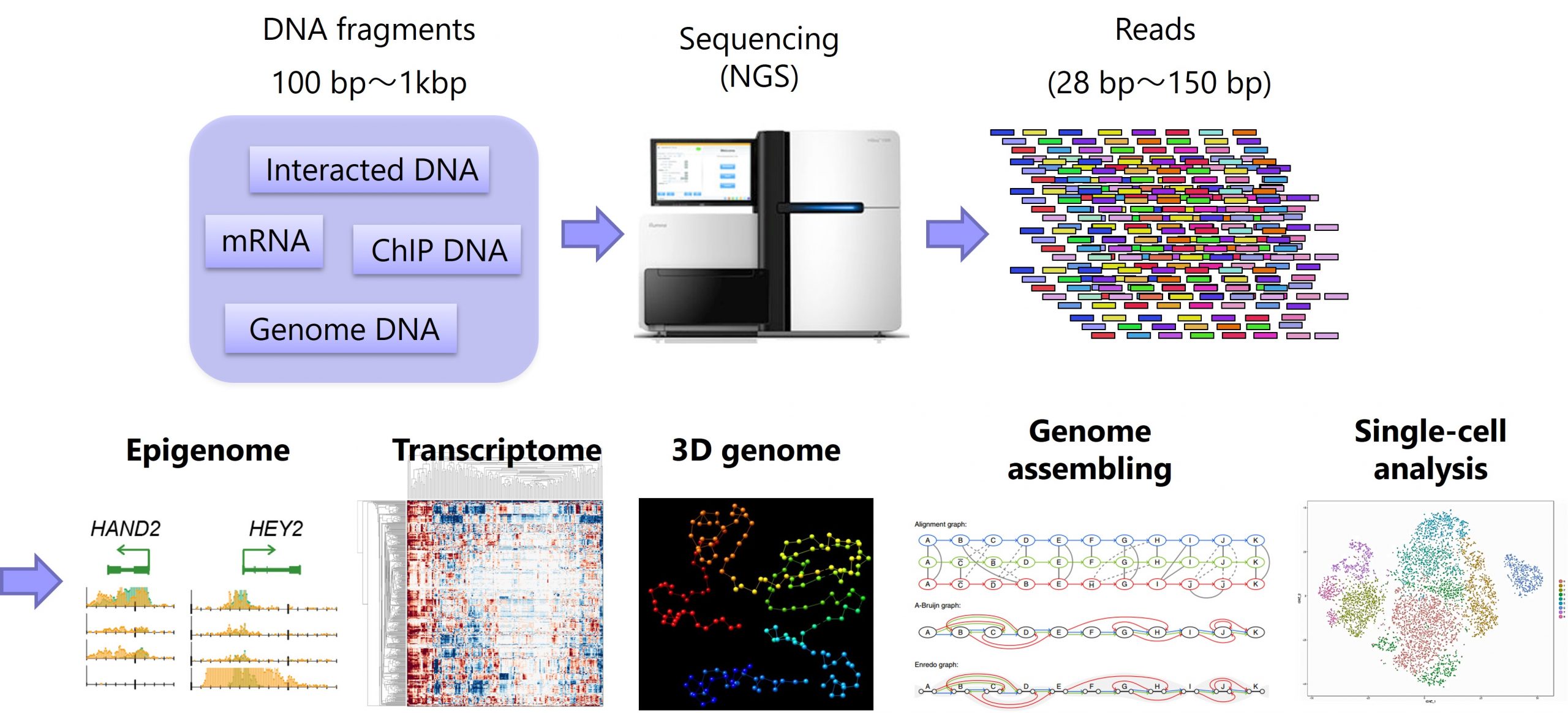
A genome of an organism contains all information for its biological activity and heredity, on which various events (e.g., gene activation, DNA replication and chromatin folding) are cooperatively regulated. Genome-wide analyses with Next Generation Sequencer (NGS) is a mainstream method in computational genomics and has led to important discoveries for dynamic regulation on the genome. We are interested in understanding the cooperative regulation of genomic events and its dysregulation in diseases by using computational genomics strategy. In collaboration with multiple wet labs, we have been investigating various cell-lines such as rare-disease patients and knockout mice by NGS assays including:
– ChIP-seq: protein-DNA binding,
– ATAC-seq: open chromatin,
– RNA-seq: gene expression,
– Exome-seq: gene mutation,
– Hi-C: 3D genome folding,
– ChIA-PET: chromatin looping, and
– Single-cell analysis: cellular heterogeneity and differential analysis.
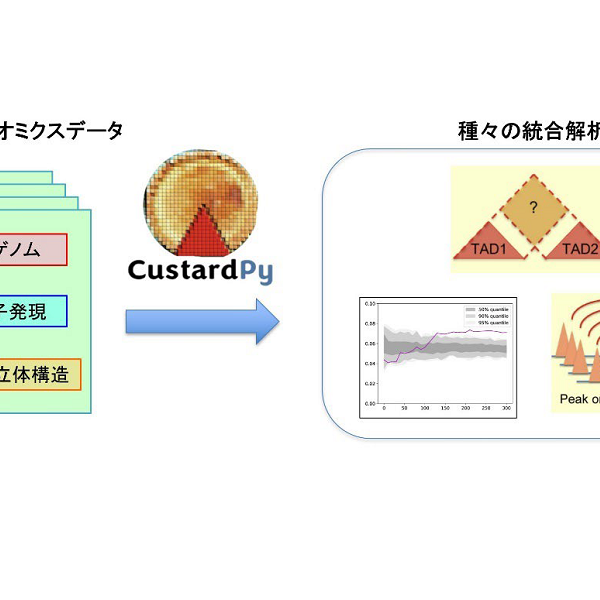
Our research focus is “data-driven NGS analysis” that extracts important biological information from large NGS datasets (~hundreds of samples) without help from existing biological knowledge. We aim to develop a pipeline for multi-NGS omics that integratively analyzes large datasets from multiple NGS assays and achieve an epoch-making discovery, e.g., higher-order coordination of multiple DNA-binding factors.
We also put effort into cultivating next-generation computational genomicists. We welcome enthusiastic and motivated students both of biology and of informatics who are interested in computational genomics.
Publication
- *Nakato R, Sakata T, Wang J, Nagai LAE, Nagaoka Y, Oba GM, Bando M, *Shirahige K. Context-dependent perturbations in chromatin folding and the transcriptome by cohesin and related factors. Nature Communications, 13(1), 2023.
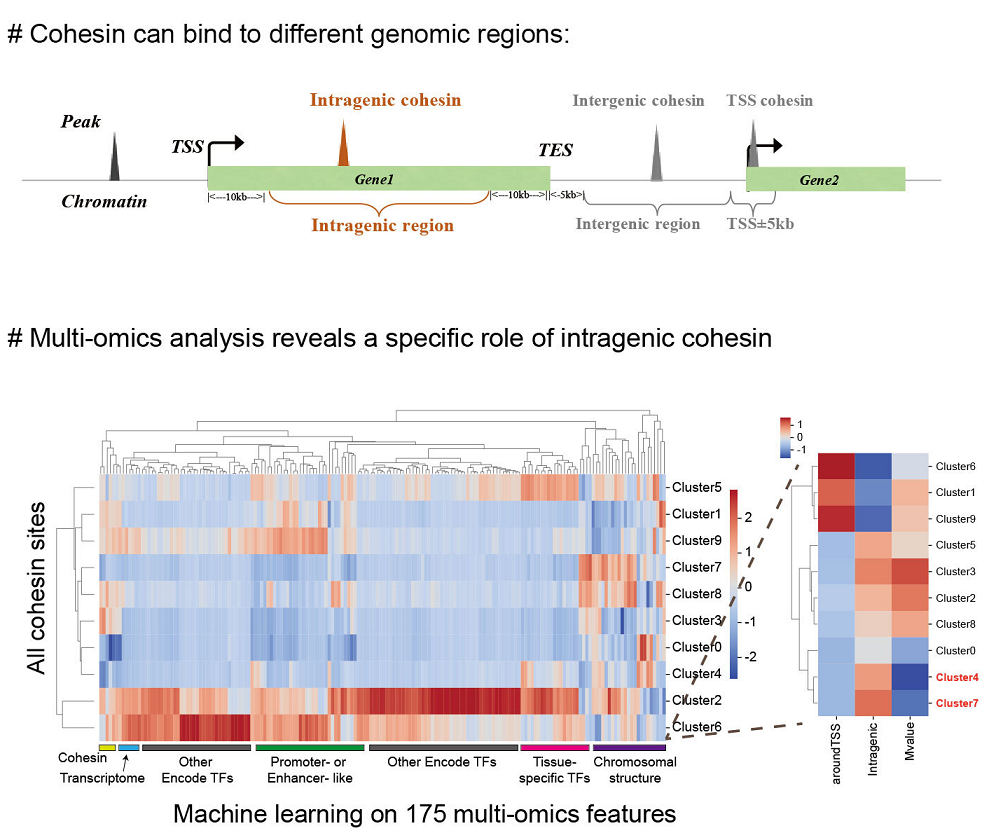
- Wang J, Bando M, Shirahige K, *Nakato R. Large-scale multi-omics analysis suggests specific roles for intragenic cohesin in transcriptional regulation. Nature Communications, 13(1), 2022.
- Wang J, *Nakato R. HiC1Dmetrics: framework to extract various one-dimensional features from chromosome structure data. Briefings in Bioinformatics, 23(1): bbab509, 2021.
- Nakajima N, Hayashi T, Fujiki K, Shirahige K, Akiyama T, Akutsu T, *Nakato R. Codependency and mutual exclusivity for gene community detection from sparse single-cell transcriptome data. Nucleic Acids Research, 49(18): e104, 2021.
- *Nakato R, Sakata T. Methods for ChIP-seq analysis: A practical workflow and advanced applications. Methods, 187: 44-53, 2020.
- ‡Nakato R, *‡Wada Y, Nakaki R, Nagae G, Katou Y, Tsutsumi S, Nakajima N, Fukuhara H, Iguchi A, Kohro T, Kanki Y, Saito Y, Kobayashi M, Izumi-Taguchi A, Osato N, Tatsuno K, Kamio A, Hayashi-Takanaka Y, Wada H, Ohta S, Aikawa M, Nakajima H, Nakamura M, McGee RC, Heppner KW, Kawakatsu T, Genno M, Yanase H, Kume H, Senbonmatsu T, Homma Y, Nishimura S, Mitsuyama T, Aburatani H, *Kimura H, *Shirahige K. Comprehensive epigenome characterization reveals diverse transcriptional regulation across human vascular endothelial cells, Epigenetics & Chromatin, 12(1): 77, 2019. ‡These authors contributed equally to this work.
- *Nakato R, Shirahige K. Sensitive and robust assessment of ChIP-seq read distribution using a strand-shift profile. Bioinformatics, 34(14): 2356-2363, 2018.
- *Nakato R, Shirahige K. Recent advances in ChIP-seq analysis: from quality management to whole-genome annotation. Briefings in Bioinformatics, 18(2): 279-290, 2017.
- Izumi K, Nakato R, Zhang Z, Edmondson AC, Noon S, Dulik MC, Rajagopalan R, Venditti CP, Gripp K, Samanich J, Zackai EH, Deardorff MA, Clark D, Allen JL, Dorsett D, Misulovin Z, Komata M, Bando M, Kaur M, Katou Y, *Shirahige K, *Krantz ID. Germline gain-of-function mutations in AFF4 cause a developmental syndrome functionally linking the super elongation complex and cohesin. Nature Genetics, 47(4): 338-344, 2015.
- *‡Deardorff MA, ‡Bando M, ‡Nakato R, ‡Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, Cole KE, De Baere E, Decroos C, Di Donato N, Ernst S, Francey LJ, Gyftodimou Y, Hirashima K, Hullings M, Ishikawa Y, Jaulin C, Kaur M, Kiyono T, Lombardi PM, Magnaghi-Jaulin L, Mortier GR, Nozaki N, Petersen MB, Seimiya H, Siu VM, Suzuki Y, Takagaki K, Wilde JJ, Willems PJ, Prigent C, Gillessen-Kaesbach G, Christianson DW, Kaiser FJ, Jackson LG, Hirota T, *Krantz ID, *Shirahige K. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature, 489(7415): 313-7, 2012. ‡These authors contributed equally to this work.
Ryuichiro Nakato
Associate Professor
Ph.D.
Graduate School of Medicine, Graduate School of Frontier Sciences
Luís Augusto Eijy Nagai
Research Associate
Ph.D.
Eiko Oikawa
Technical Specialist
Ph.D.
Naoko Yokota
Technical Specialist